Protein Misfolding
Protein folding is a complex, trial-and-error process that can sometimes result in improperly folded molecules. These misfolded proteins are usually tagged and degraded within the cell. However, this quality control system is not perfect, and intracellular or extracellular aggregates of misfolded proteins can accumulate, particularly as individuals age. Deposits of these misfolded proteins are associated with a number of diseases.
Amyloid disease
 Misfolding of proteins may occur spontaneously, or be caused by a mutation in a particular gene, which then produces an altered protein.
Misfolding of proteins may occur spontaneously, or be caused by a mutation in a particular gene, which then produces an altered protein.- In addition, some apparently normal proteins can, after abnormal proteolytic cleavage, take on a unique conformational state that leads to the formation of long, fibrillar protein assemblies consisting of β-pleated sheets.
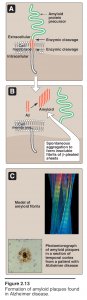
- Accumulation of these insoluble, spontaneously aggregating proteins, called amyloids, has been implicated in many degenerative diseases—particularly in the age-related neurodegenerative disorder, Alzheimer disease.
- The dominant component of the amyloid plaque that accumulates in Alzheimer disease is amyloid β (Aβ), a peptide containing 40–42 amino acid residues.
- X-ray crystallography and infrared spectroscopy demonstrate a characteristic β- pleated sheet conformation in nonbranching fibrils. This peptide, when aggregated in a β-pleated sheet configuration, is neurotoxic, and is the central pathogenic event leading to the cognitive impairment characteristic of the disease.
- The Aβ that is deposited in the brain in Alzheimer disease is derived by proteolytic cleavages from the larger amyloid precursor protein—a single transmembrane protein expressed on the cell surface in the brain and other tissues.
- The Aβ peptides aggregate, generating the amyloid that is found in the brain parenchyma and around blood vessels. Most cases of Alzheimer disease are not genetically based, although at least 5–10% of cases are familial.
- A second biologic factor involved in the development of Alzheimer disease is the accumulation of neurofibrillary tangles inside neurons. A key component of these tangled fibers is an abnormal form of the tau (τ) protein, which in its healthy version helps in the assembly of the microtubular structure. The defective τ, however, appears to block the actions of its normal counterpart.
 Misfolding of proteins may occur spontaneously, or be caused by a mutation in a particular gene, which then produces an altered protein.
Misfolding of proteins may occur spontaneously, or be caused by a mutation in a particular gene, which then produces an altered protein.Prion disease
 The prion protein (PrP) has been strongly implicated as the causative agent of transmissible spongiform encephalopathies (TSEs), including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy in cattle (popularly called “mad cow disease”).
The prion protein (PrP) has been strongly implicated as the causative agent of transmissible spongiform encephalopathies (TSEs), including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy in cattle (popularly called “mad cow disease”).- After an extensive series of purification procedures, scientists were astonished to find that the infectivity of the agent causing scrapie in sheep was associated with a single protein species that was not complexed with detectable nucleic acid.
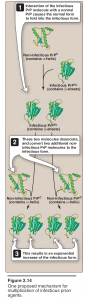
- This infectious protein is designated PrPSc (Sc = scrapie). It is highly resistant to proteolytic degradation, and tends to form insoluble aggregates of fibrils, similar to the amyloid found in some other diseases of the brain.
- A noninfectious form of PrPC (C = cellular), encoded by the same gene as the infectious agent, is present in normal mammalian brains on the surface of neurons and glial cells. Thus, PrPC is a host protein.
- No primary structure differences or alternate posttranslational modifications have been found between the normal and the infectious forms of the protein.
- The key to becoming infectious apparently lies in changes in the three-dimensional conformation of PrPC. It has been observed that a number of α-helices present in noninfectious PrPC are replaced by β-sheets in the infectious form .
- It is presumably this conformational difference that confers relative resistance to proteolytic degradation of infectious prions, and permits them to be distinguished from the normal PrPC in infected tissue.
- The infective agent is thus an altered version of a normal protein, which acts as a “template” for converting the normal protein to the pathogenic conformation. The TSEs are invariably fatal, and no treatment is currently available that can alter this outcome.
 The prion protein (PrP) has been strongly implicated as the causative agent of transmissible spongiform encephalopathies (TSEs), including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy in cattle (popularly called “mad cow disease”).
The prion protein (PrP) has been strongly implicated as the causative agent of transmissible spongiform encephalopathies (TSEs), including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy in cattle (popularly called “mad cow disease”).