Agarose Gel Electrophoresis
INTRODUCTION
For the majority of DNA samples, electrophoretic separation is carried out in agarose gels. This is because most of the DNA molecules/fragments which are routinely analyzed are larger than proteins and can not enter the polyacrylamide fine pores (except for special experiments like sequencing). Agarose gel is characterized by large pore size and is a promising medium for DNA fragments separation. The idea of using gel electrophoresis to analyze DNA was first of all given by Vin Thorne in 1966-67 to separate superhelical, nicked, and linear forms of polyomavirus DNA, radiolabeled with H3 thymidine. Since then agarose gel electrophoresis of DNA has metamorphosized from a very simple form to a very skilled and sophisticated technique.
The movement of DNA through Agarose gel is characterized by the following points:
(i) At low voltage, the rate of electrophoretic migration of linear DNA fragments is proportional to the voltage applied. But at higher voltage, the effective range of separation of fragments decreases.
(ii) Smaller molecules move faster as compared to larger molecules because of the lesser frictional force
(iii) The concentration of agarose gels also play an important role in deciding the rate of migration of DNA samples, depicted by the following equation:
Log λ = logλ-KrT
λ = Free electrophoretic mobility of DNA
Kr = Retardation coefficient
T = Concentration of gel
(iv) If the DNA is having some intercalating dyes, e.g., ethidium bromide, the electrophoretic mobility of linear DNA is reduced by approximately 15-20%.
(v) The electrophoretic mobility of DNA is affected by the composition and ionic strength of the electrophoresis buffer. Several different buffers are available for electrophoresis of native double-stranded DNA, e.g., Tris-acetate (TAE), Tris-borate (TBE), or Tris phosphate (TPE) at a concentration of approximately 50 mM (pH 7.5-7.8). TPE and TBE are slightly more expensive than TAE, but they have significantly higher buffering capacity. Double-stranded linear DNA fragments migrate approximately 10% faster through TAE than through TBE or TPE but the resolving power of these systems are almost identical.
MATERIALS/REAGENTS REQUIRED
1. Agarose solutions (1%)
Take 1 gm of agarose in a conical flask, add 100 ml of 1 X electrophoresis buffer, and set for melting until a clear transparent solution is achieved.
2. 10 X TBE
| Tris base Boric acid 0.5 M EDTA (pH 8.0) Add water to 1 Litre | 108 g 55 g 40 ml |
3. 1 X TBE
Take 10 ml of 10 x TBE and dilute to 100 ml with double distilled water.
4. Ethidium bromide (10 mg/ml in sterile double distilled water)
5. 10 X gel loading dye
| Glycerol 10 X TBE Bromophenol blue (Saturated) Xylene cyanol 10% Mix well, autoclave, and store in aliquots. | 5 ml 1 ml 1 ml 1 ml
|
6. DNA samples
7. DNA size standards
8. Horizontal slab gel electrophoresis apparatus
9. Gel-sealing tape
10. Microwave oven
11. Power supply device capable of up to 500 V and 200 mA
12. Water bath preset to 55°C
13. UV Transilluminator
Experimental Layout
METHOD
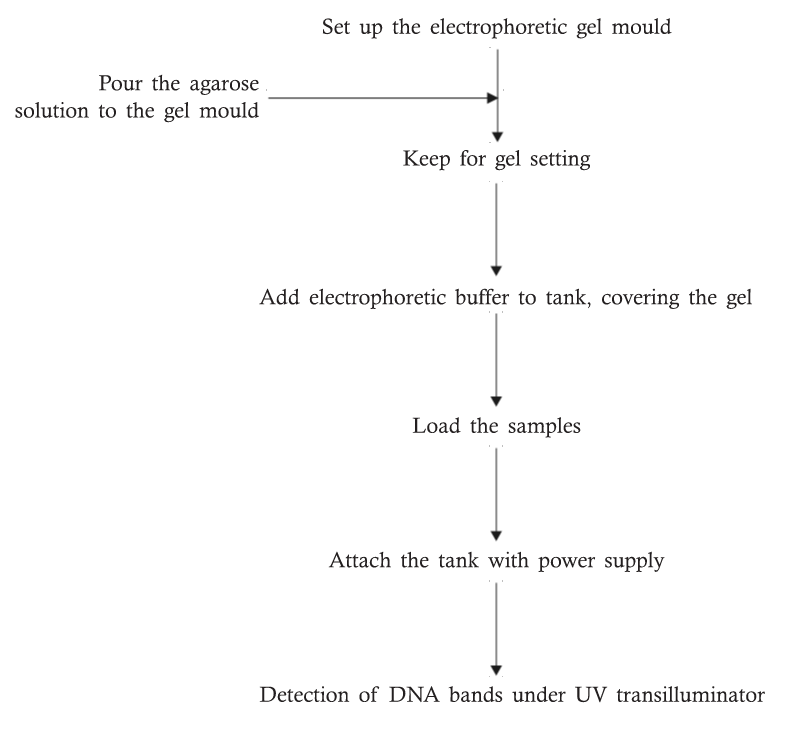
- Prepare a 1% solution of agarose by adding 100 ml of 1 X TBE buffer to 1 gm of weighed agarose in a 250 ml conical flask.
- Melt the agarose completely in a clear solution, using a microwave oven.
- When melted, place the flask having melted agarose in a water bath set at 55°C until it is cool enough to handle.
- While the agarose is melting, tightly seal the end of the gel tray with plastic tape. After that keep the comb in position.
- When the melted agarose is cool enough (up to 50°C), add 10 ul of ethidium bromide (10 mg/ml) and mix well.
- Pour the cooled gel solution into the gel mold and allow the gel to set for 20 minutes.
- When the gel hardens, remove the tape and comb. Place get tray into the box. Add buffer to the tank so that it covers the gel to a depth of about 2 mm.
- Load the DNA samples carefully into the wells. In one well load a DNA size standard.
- Close the lid of the gel tank and attach the electrical leads so that the DNA will migrate toward the positive end (red lead). Apply a voltage of 50 volts, bubbles will be generated at the electrodes within a few minutes and the bromophenol blue will migrate from the wells into the body of the gel.
- Stop electrophoresis, when bromophenol blue crosses more than 2/3 of the length of the gel (in no case it should move out of the gels).
- Visualize DNA bands on a UV trans-illuminator.
- If required, this profile can be photographed through a red filter for documentation.
REMEMBER
- 1% as used in the above protocol, is the standard concentration, but if you are trying to resolve large fragments of DNA, you may use a lower concentration and for small fragments, a higher concentration would be appropriate.
- Adding the ethidium bromide directly to the gel is an optional step. If you don’t wish to add the dye here, you will have to stain the gel at the end. It is sometimes useful to have the dye in the gel while the gel is running because you can also interrupt the run, check the location of your DNA fragments, and then continue if you wish to run further. But ethidium bromide alters the conformation of the DNA, thereby altering the migration rate. So if you are to generate a restriction map and would like to measure fragment sizes accurately, it is always preferable to run the gel in the absence of the dye. In this case, a brief destaining either with water or 1 mM magnesium sulfate solution is necessary.
Caution: Ethidium bromide is a powerful carcinogen. Take all the precautions, while handling.