What is Hemoglobin?
- Definition: Hemoglobin is a metalloprotein in red blood cells responsible for transporting oxygen from the lungs to tissues and facilitating the return of carbon dioxide to the lungs for exhalation.
- Location: Primarily found in erythrocytes (red blood cells).
- Significance: Essential for oxygen delivery, carbon dioxide transport, and buffering blood pH, making it vital for cellular respiration and metabolic homeostasis.
- Composition: A tetrameric protein consisting of globin polypeptide chains and heme prosthetic groups.
Structure of Hemoglobin
Hemoglobin’s structure is intricately designed to perform its functions efficiently. Below, we explore its components, organization, and conformational changes.
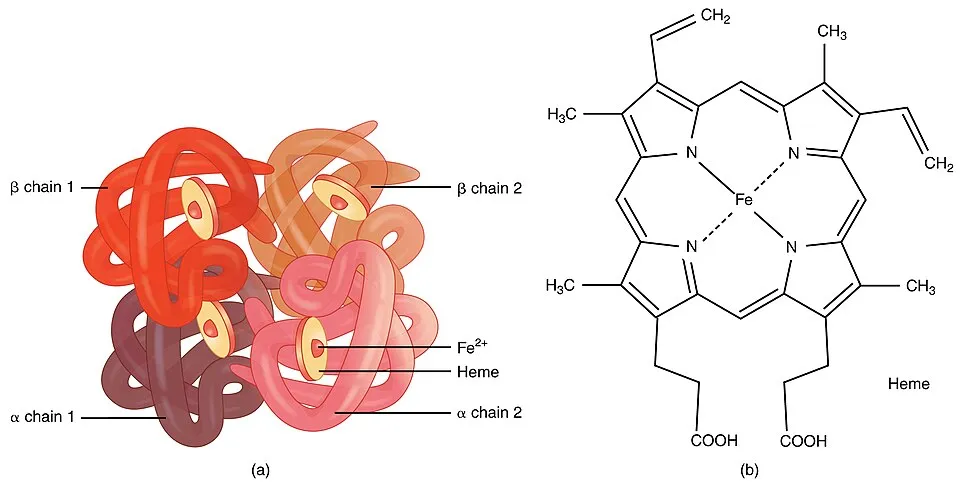
Components of Hemoglobin
- Heme Group:
- A porphyrin ring complex containing a central iron (Fe²⁺) atom.
- The iron atom binds oxygen reversibly, enabling oxygen transport.
- Each heme group is embedded in a globin chain.
- Globin Chains:
- Polypeptide chains that provide structural support to the heme group.
- Adult hemoglobin (HbA) consists of two alpha (α) and two beta (β) chains, forming a tetramer (α₂β₂).
- Quaternary Structure:
- Hemoglobin is a tetrameric protein with four subunits (two α and two β chains).
- Subunits are held together by non-covalent interactions, including hydrogen bonds, van der Waals forces, and hydrophobic interactions.
Types of Hemoglobin
- Adult Hemoglobin (HbA): Comprises 97–98% of hemoglobin in adults, with the structure α₂β₂.
- Fetal Hemoglobin (HbF): Found in fetuses, composed of two alpha (α) and two gamma (γ) chains (α₂γ₂). Has a higher oxygen affinity to extract oxygen from maternal blood.
- Hemoglobin A2 (HbA2): Minor adult hemoglobin (2–3%), composed of two alpha (α) and two delta (δ) chains (α₂δ₂).
- Embryonic Hemoglobin: Found in early embryos, includes types like Gower 1 (ζ₂ε₂), Gower 2 (α₂ε₂), and Portland (ζ₂γ₂).
Table 1: Types of Hemoglobin and Their Composition
| Hemoglobin Type | Chain Composition | Location/Function | Oxygen Affinity |
| HbA (Adult) | α₂β₂ | Adult RBCs | Moderate |
| HbF (Fetal) | α₂γ₂ | Fetal RBCs | High |
| HbA2 | α₂δ₂ | Adult RBCs (minor) | Moderate |
| Gower 1 | ζ₂ε₂ | Early embryo | High |
| Gower 2 | α₂ε₂ | Early embryo | High |
| Portland | ζ₂γ₂ | Early embryo | High |
Structural Features
- Heme Binding Pocket:
- The iron atom in the heme group is coordinated to four nitrogen atoms in the porphyrin ring and a histidine residue (proximal histidine) in the globin chain.
- A distal histidine stabilizes oxygen binding but prevents oxidation of Fe²⁺ to Fe³⁺.
- Cooperative Binding:
- Hemoglobin exhibits cooperative oxygen binding, where binding of oxygen to one heme group increases the affinity of other heme groups for oxygen (allosteric effect).
- This is described by the sigmoid-shaped oxygen dissociation curve.
- Conformational States:
- T (Tense) State: Deoxygenated hemoglobin with low oxygen affinity.
- R (Relaxed) State: Oxygenated hemoglobin with high oxygen affinity.
- Transition between T and R states is facilitated by oxygen binding, modulated by allosteric effectors like 2,3-bisphosphoglycerate (2,3-BPG), H⁺, and CO₂.
Table 2: Structural Features of Hemoglobin
| Feature | Description |
| Heme Group | Porphyrin ring with Fe²⁺, binds oxygen reversibly |
| Globin Chains | Two α and two β chains in HbA, provide structural support |
| Cooperative Binding | Oxygen binding to one subunit increases affinity in others |
| T State | Deoxygenated, low oxygen affinity, stabilized by 2,3-BPG |
| R State | Oxygenated, high oxygen affinity, stabilized by oxygen binding |
Allosteric Effectors
- 2,3-Bisphosphoglycerate (2,3-BPG):
- Binds to the central cavity of deoxygenated hemoglobin, stabilizing the T state and reducing oxygen affinity.
- Facilitates oxygen release in tissues with high metabolic demand.
- Carbon Dioxide (CO₂):
- Binds to hemoglobin, forming carbaminohemoglobin, which stabilizes the T state.
- Protons (H⁺):
- Low pH (high H⁺ concentration) reduces oxygen affinity (Bohr effect), promoting oxygen release in acidic tissues.
- Carbon Monoxide (CO):
- Binds to heme with 200–250 times higher affinity than oxygen, forming carboxyhemoglobin, which is toxic.
Synthesis of Hemoglobin
Hemoglobin synthesis is a tightly regulated process occurring primarily in erythroid precursor cells in the bone marrow. It involves the production of heme and globin chains, followed by their assembly.
Site of Synthesis
- Location: Bone marrow, specifically in erythroblasts (immature red blood cells).
- Cell Types: Reticulocytes (immature RBCs) also contribute to hemoglobin synthesis during final maturation stages.
Steps in Hemoglobin Synthesis
- Heme Synthesis:
- Location: Mitochondria and cytoplasm of erythroid cells.
- Key Enzyme: Aminolevulinic acid synthase (ALAS), which catalyzes the formation of 5-aminolevulinic acid (ALA) from glycine and succinyl-CoA.
- Process:
- ALA is synthesized in mitochondria and transported to the cytoplasm.
- Two ALA molecules condense to form porphobilinogen (PBG).
- PBG molecules form a tetrapyrrole ring, which is modified to produce protoporphyrin IX.
- Iron (Fe²⁺) is inserted into protoporphyrin IX by ferrochelatase in mitochondria, forming heme.
- Regulation: Heme inhibits ALAS to prevent overproduction (negative feedback).
- Globin Chain Synthesis:
- Location: Cytoplasmic ribosomes.
- Genes Involved:
- Alpha-globin genes on chromosome 16.
- Beta-globin genes on chromosome 11.
- Process:
- Transcription of globin genes produces mRNA.
- mRNA is translated into α and β polypeptide chains.
- Chaperone proteins ensure proper folding of globin chains.
- Regulation: Balanced production of α and β chains is critical to prevent excess chain accumulation, which can lead to disorders like thalassemia.
- Assembly of Hemoglobin:
- Heme is transported from mitochondria to the cytoplasm.
- Heme binds to globin chains, forming a heme-globin complex.
- Two α and two β chains combine with four heme groups to form the tetrameric hemoglobin molecule.
- Quality control mechanisms ensure proper folding and assembly.
Table 3: Key Steps in Hemoglobin Synthesis
| Stage | Location | Key Components/Processes |
| Heme Synthesis | Mitochondria, Cytoplasm | ALA → PBG → Protoporphyrin IX → Heme |
| Globin Synthesis | Cytoplasmic Ribosomes | Transcription and translation of α and β genes |
| Hemoglobin Assembly | Cytoplasm | Heme + Globin → Tetramer (α₂β₂) |
Regulation of Hemoglobin Synthesis
- Heme Feedback Inhibition: Excess heme inhibits ALAS to regulate heme production.
- Iron Availability: Iron deficiency impairs heme synthesis, leading to anemia.
- Erythropoietin (EPO): A hormone produced by the kidneys that stimulates erythroid cell proliferation and hemoglobin synthesis in response to low oxygen levels.
- Transcription Factors: GATA-1 and EKLF regulate globin gene expression.
- Thalassemia Control: Balanced α and β chain production prevents precipitation of excess chains, which can damage RBCs.
Breakdown of Hemoglobin
Hemoglobin breakdown occurs when red blood cells reach the end of their lifespan (approximately 120 days). The process is carried out primarily in the spleen, liver, and bone marrow.
Overview of Hemoglobin Catabolism
- Purpose: To recycle iron, degrade heme, and eliminate waste products.
- Key Organs:
- Spleen: Primary site for RBC destruction by macrophages.
- Liver: Processes hemoglobin breakdown products.
- Bone Marrow: Recycles iron for new RBC production.
Steps in Hemoglobin Breakdown
- RBC Phagocytosis:
- Senescent or damaged RBCs are engulfed by macrophages in the spleen’s red pulp.
- Hemoglobin is released and broken down into heme and globin.
- Globin Degradation:
- Globin chains are hydrolyzed into amino acids by proteases.
- Amino acids are recycled for protein synthesis or metabolized for energy.
- Heme Degradation:
- Enzyme: Heme oxygenase in macrophages.
- Process:
- Heme is cleaved to release iron (Fe²⁺), carbon monoxide (CO), and biliverdin.
- Iron is bound to transferrin for transport to bone marrow or stored as ferritin.
- Biliverdin is reduced to bilirubin by biliverdin reductase.
- Bilirubin Processing:
- Unconjugated bilirubin is transported to the liver bound to albumin.
- In the liver, bilirubin is conjugated with glucuronic acid by UDP-glucuronosyltransferase, forming conjugated bilirubin.
- Conjugated bilirubin is excreted into bile and released into the intestine.
- In the gut, bacteria convert bilirubin to urobilinogen, which is further oxidized to urobilin (excreted in urine) or stercobilin (excreted in feces).
Table 4: Hemoglobin Breakdown Pathway
| Stage | Location | Key Processes |
| RBC Phagocytosis | Spleen (Macrophages) | RBCs engulfed, hemoglobin released |
| Globin Degradation | Macrophages | Globin → Amino acids |
| Heme Degradation | Macrophages | Heme → Iron + CO + Biliverdin → Bilirubin |
| Bilirubin Processing | Liver, Intestine | Conjugation → Excretion as urobilin/stercobilin |
Recycling and Waste Elimination
- Iron Recycling:
- Iron is transported by transferrin to bone marrow for new hemoglobin synthesis.
- Excess iron is stored as ferritin in the liver or spleen.
- Carbon Monoxide: Exhaled via the lungs.
- Bilirubin Byproducts:
- Urobilin gives urine its yellow color.
- Stercobilin gives feces its brown color.
Clinical Relevance
- Jaundice: Accumulation of bilirubin due to impaired liver function or excessive RBC breakdown.
- Hemolytic Anemia: Accelerated RBC destruction leads to increased hemoglobin breakdown and bilirubin production.
- Iron Deficiency: Inadequate iron recycling or dietary intake impairs hemoglobin synthesis.
Clinical and Physiological Importance
- Oxygen Transport: Hemoglobin’s ability to bind and release oxygen ensures efficient delivery to tissues.
- Carbon Dioxide Transport: Hemoglobin carries ~20% of CO₂ as carbaminohemoglobin, aiding in CO₂ removal.
- pH Buffering: Hemoglobin buffers blood pH by binding H⁺ ions, maintaining acid-base balance.
- Disorders:
- Anemia: Low hemoglobin levels due to iron deficiency, blood loss, or impaired synthesis.
- Thalassemia: Imbalance in α and β chain production, leading to defective hemoglobin.
- Sickle Cell Disease: Mutation in the β-globin gene (HbS) causes abnormal hemoglobin, leading to RBC sickling.
- Methemoglobinemia: Oxidation of Fe²⁺ to Fe³⁺ impairs oxygen binding.
Table 5: Clinical Disorders Related to Hemoglobin
| Disorder | Cause | Symptoms |
| Anemia | Low hemoglobin (iron deficiency, blood loss) | Fatigue, pallor, shortness of breath |
| Thalassemia | Imbalanced α/β chain production | Anemia, bone deformities, organ damage |
| Sickle Cell Disease | HbS mutation (β-globin) | Pain, infections, organ damage |
| Methemoglobinemia | Fe²⁺ oxidized to Fe³⁺ | Cyanosis, shortness of breath |
FAQs
- What is the primary function of hemoglobin?
- Hemoglobin transports oxygen from the lungs to tissues and carries carbon dioxide back to the lungs for exhalation.
- How is hemoglobin synthesized?
- Hemoglobin synthesis occurs in bone marrow erythroblasts, involving heme production in mitochondria and globin synthesis on ribosomes, followed by assembly into a tetramer.
- What happens to hemoglobin when red blood cells die?
- Hemoglobin is broken down in the spleen by macrophages. Globin is degraded into amino acids, heme is converted to bilirubin, and iron is recycled.
- What is the difference between adult and fetal hemoglobin?
- Adult hemoglobin (HbA) has α₂β₂ chains with moderate oxygen affinity, while fetal hemoglobin (HbF, α₂γ₂) has higher oxygen affinity to extract oxygen from maternal blood.
- Why does jaundice occur?
- Jaundice results from excessive bilirubin accumulation due to increased RBC breakdown or impaired liver processing.