Nucleic Acid Hybridization
BASIC CONSIDERATIONS
Nucleic acid hybridization is a basic tool in molecular genetics that exploits the ability of single-stranded nucleic acid molecules to hybridize with complementary sequences to form double-stranded molecules. The principle of hybridization, in general, is the addition of a probe to a complex mixture of target DNA. The mixture is incubated under conditions that promote the formation of hydrogen bonds between complementary strands. The factors that affect hybridization characteristics are:
- Strand Length-The longer the probe the more stable the duplex
- Base Composition— The % G:C base pairs are more stable than A:T
- Chemical environment—The concentration of Na+ ions stabilizes.
Chemical denaturants (formamide or urea) destabilize hydrogen bonds.
Therefore, in nucleic acid hybridization, single-stranded nucleic acids (DNA or RNA) are allowed to interact so that complexes (hybrids) are formed by molecules with complementary sequences. Through nucleic acid hybridization, the degree of sequence identity between nucleic acids can be determined and specific sequences can be detected. The most important factor in nucleic acid hybridization is the high degree of base complementarity. In nucleic acid hybridization assay systems, a labeled nucleic acid molecule (probe) is used to identify a complementary nucleic acid (DNA or RNA) molecule amongst a mixture of unlabelled nucleic acid molecules. The labeled nucleic acid molecule is known as probe and the nucleic acid sequence identified by the labeled nucleic acid molecule (because of complementarity) is known as target nucleic acid. Nucleic acid hybridizations can be done in all combinations: DNA-DNA (DNA can be rendered single-stranded by denaturation), DNA-RNA, or RNA-RNA. In situ hybridization involves hybridizing a labeled nucleic acid to prepared cells or histological sections. This is used particularly to look for specific transcription/localization of genes to specific chromosomes via fluorescent in situ hybridization (FISH) analysis. Nucleic acid hybridization is used in cloning and PCR also.
Nucleic Acid Hybridization Method- This method was developed in 1975 by Grunstein and Hogness. It is used to determine the colony which contains the sequence from hundreds of clones. In this method, bacterial colonies are transferred from the agar plate onto the nitrocellulose membrane and lysed by alkaline solution. After the bacterial cell wall lysis, the proteins and DNA adhere to the nitrocellulose membrane because of the membrane’s negative charge. Then the membrane is placed in proteinase K solution to remove the protein bound to the membrane and DNA. Then DNA is exposed to UV rays so that the DNA gets fixed onto the nitrocellulose membrane. After baking, the membrane is exposed to labeled RNA for hybridization. Then RNA-DNA hybrids are monitored by autoradiography. A colony which gives a positive result can then be picked from the master plate.
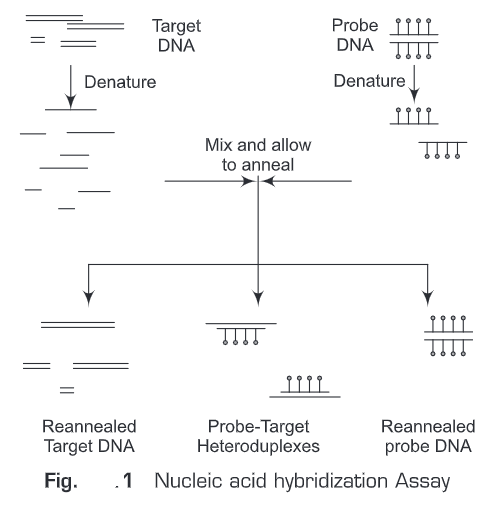
In general, nucleic acid hybridization method (Fig. 1) involves the following steps:
1. A nucleic acid probe is needed which will anneal to the target nucleic acid
2. The target is attached to a solid matrix e.g., membrane
3. Both the probe and target are denatured
4. Denatured probe is added to the target in a solution
5. If there is sequence homology between the target and the probe, the probe will hybridize/ anneal to the target
6. The hybridized probe is the detected by autoradiography, chemiluminescence or colorimetric
A variety of techniques utilize hybridization of DNA or RNA probes. Such as:
- ASO
- Southern Blot
- RFLP
- VNTRs
- Northern Blot
- tissue specific expression
- In situ hybridization
- Chromosome location and integrity
- Tissue specific expression
HYBRIDIZATION PROBES
A hybridization probe is a nucleic-acid (DNA or RNA) fragment that is complementary to another nucleic-acid sequence and thus, when labeled (with radioisotope, fluorescent dye, etc) can be used to identify complementary segments present in the nucleic-acid sequences of various microorganisms. The probe actually hybridizes to single-stranded nucleic acid (DNA or RNA) molecules because of complementarity between the probe and target. The labeled probe is first denatured into single DNA strands and then hybridized to the target DNA (Southern blotting) or RNA (northern blotting) immobilized on a membrane or in situ. To detect probe hybridization to its target sequence, the probe is labeled with a molecular marker (radioactive -32P, a radioactive isotope of phosphorus incorporated into the phosphodiester bond in the probe DNA; or fluorescent molecules – Digoxigenin).
Nucleic acid probes are synthesized in the laboratory. Nucleic acid probes can be synthesized as single-stranded or double-stranded probes, but a working nucleic acid probe should be single-stranded only (so as to bind with complementary target sequence).
Nucleic acid hybridization probes (Fig. 2) are of three types:
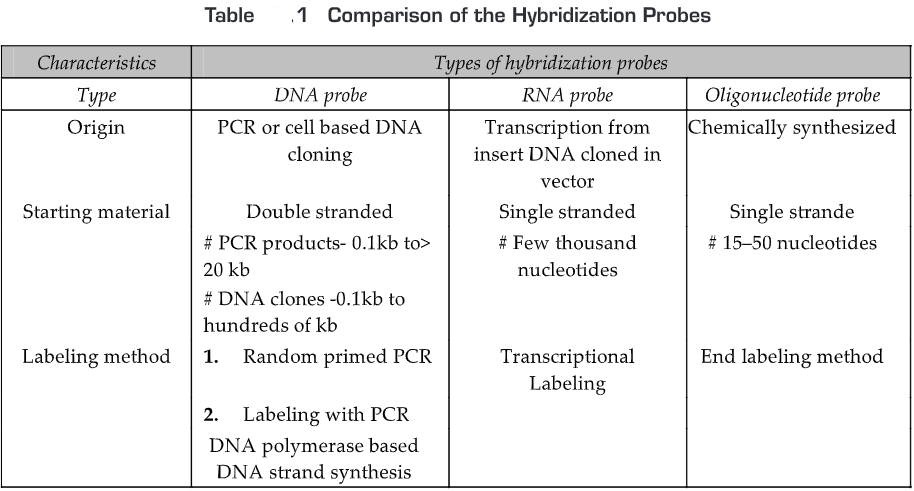
DNA Probe
A single-stranded DNA molecule used in laboratory experiments to detect the presence of a complementary sequence among a mixture of other singled-stranded nucleic acid molecules. Therefore, DNA probe is a short sequence of DNA labeled isotopically or chemically that is used for the detection of a complementary nucleotide sequence.
RNA Probe (Riboprobes)
A single-stranded RNA molecule used in laboratory experiments to detect the presence of a complementary sequence among a mixture of other singled-stranded nucleic acid molecules. Therefore, RNA probe is a short sequence of RNA labeled isotopically or chemically that is used for the detection of a complementary nucleotide sequence. Single-stranded RNA probes also called complementary RNA (cRNA) or riboprobes and are often used for in situ hybridization because: of high sensitivity; RNA-RNA hybrids are more stable than DNA-RNA hybrids; and non-specific tissue signals can be removed after hybridization with RNase A since RNA duplexes are resistant to degradation by RNase A.
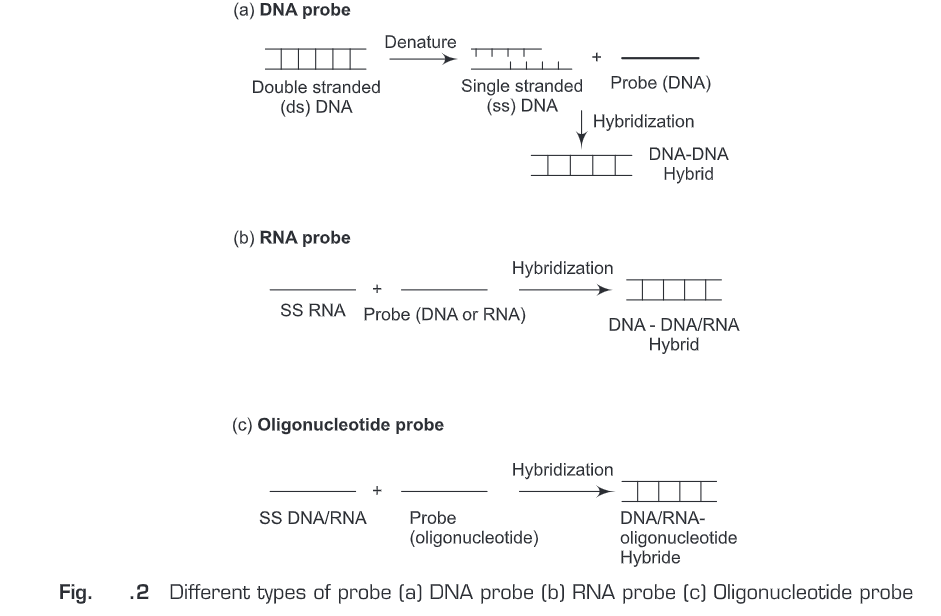
Oligonucleotide Probe
Oligonucleotide probe is a short sequence of nucleotides synthesized to match a region where a mutation is known to occur and then used as a molecular probe to detect the mutation. Oligonucleotide probes are synthesized chemically in the laboratory. Initially, a mononucleotide is bound to a solid support and then more mononucleotides are added one by one to the 3’end of the membrane-bound mononucleotide. Oligonucleotides are generally labeled by adding a marker molecule (radioactive or fluorescent dye) at the 5’end.
The comparison of the hybridization probes is given in Table 1.
LABELING OF HYBRIDIZATION PROBES
Hybridization probes can be labeled by two methods (Fig. 3)
In Vivo Labeling
By supplying labeled nucleotides to the cultured cells.
In Vitro Labeling
An enzyme is used to incorporate a labeled nucleotide in the probe
(a) Strand synthesis labeling for DNA probes
(i) Labeling by nick translation
(ii) Random primed labeling
(iii) PCR mediated labeling
(b) Strand synthesis labeling for RNA probes
(i) In vitro transcription from cloned DNA inserts
(ii) 3’-end labeling RNA
(c) End labeling for oligonucletide probes
(i) Kinase end labeling
(a) Fill-in end labeling
(iii) Primer mediated 5’end labeling
(a) Strand synthesis labeling for DNA probes
Strand synthesis labeling is the most common labeling method for DNA. In this method DNA polymerase enzyme is used to incorporate labeled nucleotides in the DNA copies of the starting DNA. Any one of the nucleotides is labeled and is then incorporated in the DNA synthesis reaction mixture. Labeling of DNA is generally done by any one of the following methods:
(i) Labeling by nick translation— Nick translation (Fig. 4) is a procedure for making a DNA probe in which a DNA fragment is treated with DNase to produce single-stranded nicks, followed by incorporation of labeled nucleotides from the nicked sites by DNA polymerase I. This process is called nick translation because the DNA to be processed is treated with DNase to produce single-stranded “nicks.” This is followed by replacement in nicked sites by DNA polymerase I, which elongates the 3′ hydroxyl terminus, removing nucleotides by 5′-3′ exonuclease activity, replacing them with labeled dNTPs. This nick is then sealed by DNA ligase. If the reaction is carried at 15°C (low temperature), the reaction will renew only one copy of the original nucleotide strand. The advantage of this method is that it allows the control of probe length which is important in situ hybridization applications.

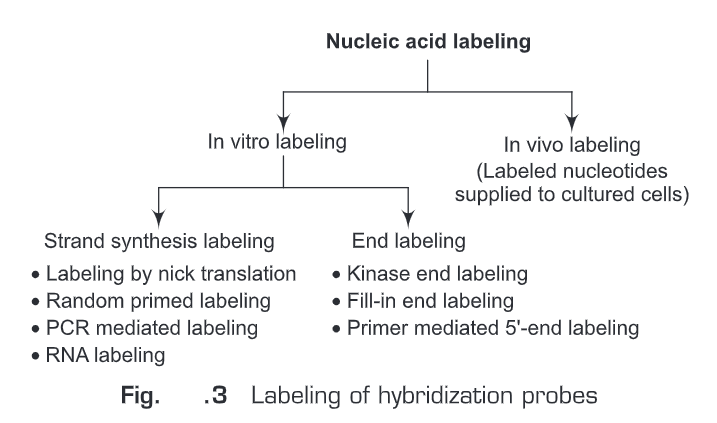
(ii) Random primed labeling —The method of “random primed” DNA labeling was introduced by Feinberg and Vogelstein in 1983. This method is based on the hybridization of oligonucleotides of all possible sequences to the denatured template DNA to be labeled. The complementary DNA strand is synthesized by a “Klenow” fragment of DNA Polymerase I, using the random oligonucleotides as primers. A labeled nucleotide is added in the reaction mixture. Therefore, the newly synthesized complementary DNA will be labeled. The reaction mixture will contain random oligonucleotides, a Klenow fragment of DNA Polymerase I, dATP, dGTP, dTTP. This rapid labeling is accomplished with the use of the Klenow fragment, which lacks 5′-3′ exonuclease activity, and by the use of hexamer/nonamer primers giving more efficient priming from the template at 37°C (Fig. 5).
Random primed labeling method produces labeled DNA of high specific activity. The advantages of this method are:
- It produces very sensitive probes
- It can be scaled up tenfold
- It can label templates of almost any length
(iii) PCR mediated labeling— Amplification of DNA using the polymerase chain reaction (PCR) provides the opportunity to label the resulting product with either modified nucleotides or oligonucleotide primers. Therefore, we can incorporate either the labeled nucleotides in the reaction mixture or the labeled primers in the PCR reaction mixture which will result in the production of labeled PCR product throughout its length. PCR labeling produces a very high yield of labeled probe from very little template. The advantages of this method are:
- It requires only a small amount of template
- Impure templates can be used
- It requires less optimization than other methods
- Has high yield of labeled probe
- It is recommended for very short probes (<100 bp)
- It produces very sensitive probes
(b) Strand synthesis labeling for RNA probes
Strand synthesis labeling is a common labeling method for RNA. In this method RNA polymerase enzyme is used to incorporate labeled nucleotides in the RNA copies of the starting RNA. Anyone of the nucleotides is labeled and is then incorporated in the RNA synthesis reaction mixture.
Labeling of RNA is done in dark by:
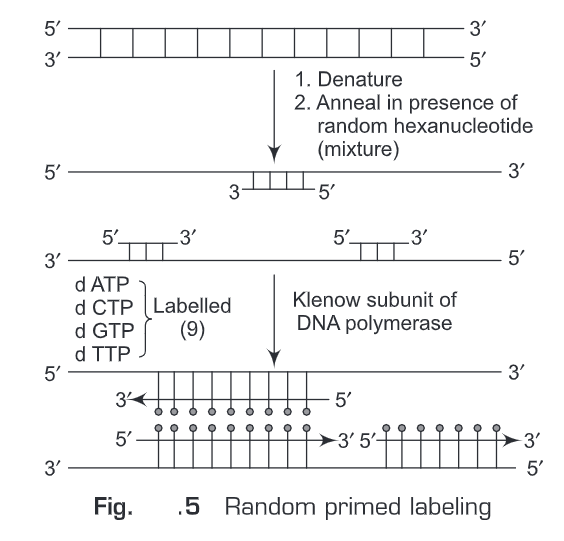
(i) In vitro transcription from cloned DNA inserts—RNA probes are prepared by in vitro transcription (Fig. 6). The RNA probe is transcribed from a linear DNA template using bacteriophage DNA-dependent RNA polymerases from the Salmonella bacteriophage SP6, and the E. coli. bacteriophages T3 and T7 (RNA polymerase T7, T3 or SP6). We should have sufficient quantities of a plasmid carrying the gene sequence of interest that can be used as the template for RNA probe synthesis. Before a riboprobe can be transcribed, the correct RNA polymerase promoter sequences must be available in the plasmid in the correct orientation with respect to the template sequence. The transcription vectors pGEM (SP6 and T7 promoters) and pBluescript (T3 and T7 promoters) are commonly used. For in vitro transcription reactions the plasmid must also be in a linear form. We use restriction enzymes to linearize the plasmid. For example, the plasmid vector (pSP64) contains SP6 promoter sequence adjacent to multiple cloning sites (MCS). Then transcription is initiated from a specific start point in the promoter sequence transcribing the DNA sequence that has been inserted in the multiple cloning sites (MCS). Labeled sense and anti-sense riboprobes can be generated and are widely used in tissue in situ hybridization.
The advantages of this method are :
- It generates large amounts of probe
- Labeled probe is completely free of vector sequences
- RNA probes are single-stranded and cannot re-anneal to an opposite strand
- RNA probes are more sensitive than DNA probes for analyzing northern blots
- DIG-labeled RNA probes can easily be fragmented for in situ hybridization
(ii) 3’-end labeling RNA – A short DNA template is designed to anneal to the 3’-end of the RNA, with a two nucleotide 5’ overhang of 3’-TA-5’, 3’-TG-5’ or 3’-TC-5’. The Klenow fragment of DNA polymerase I can then cleanly and efficiently extend the 3’-end of the RNA by the incorporation of a single a-32P-labeled dATP residue. This method can be used to label one RNA ina mixture of RNAs, or to label 5’-blocked RNAs such as mRNA.
(c) End labeling for oligonucleotide probes
Oligonucleotide probes are sensitive enough to detect single copy gene sequences in complex genomes if sufficient target DNA (e.g. 10 ug human genomic DNA) is present on the blot. For some applications, such as in situ hybridization, a labeled synthetic oligonucleotide is the best hybridization probe. In addition to in situ hybridizations, labeled oligonucleotides may be used as hybridization probes in
- Dot/slot blots
- Library screening
- Detection of repeated gene sequences on Southern blots
- Detection of abundant mRNAs on Northern blots
Advantages of oligonucleotide probes are:
Oligonucleotides are small and diffuse readily into cells, making them ideal for in situ
hybridization applications
- Small probe hybridizes to target faster, so hybridization times are reduced
- Oligonucleotide probes are single-stranded, so they cannot self-hybridize (re-nature) during the hybridization reaction
- Oligonucleotide sequences can be custom synthesized, so target recognition is optimized.
Methods used for labeling of oligonucleotides are :
(i) Kinase end labeling
(ii) Fill-in end labeling
(iii) Primer mediated 5’end labeling
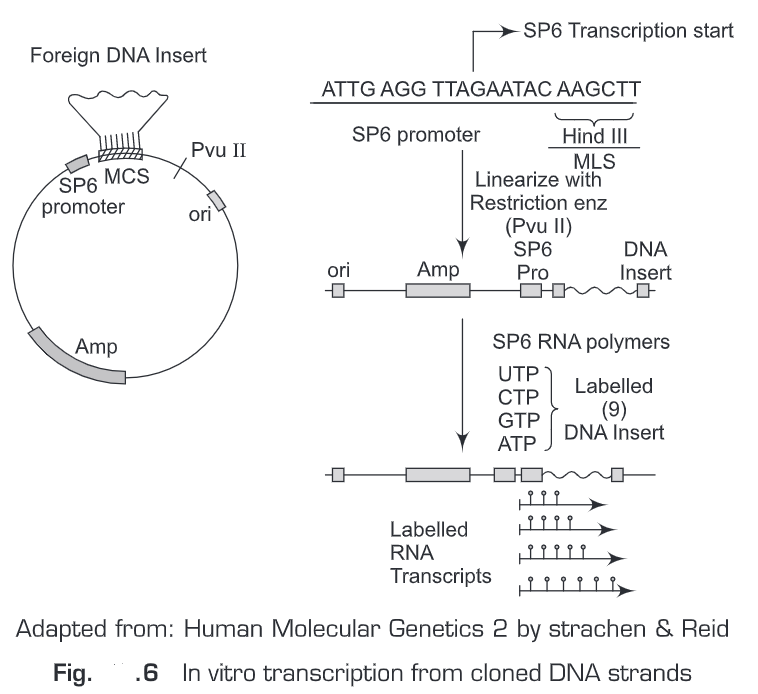
(i) Kinase end labeling—This is the most common method for oligonucleotide labeling. Polynucleotide kinase (therefore named kinase end labeling) is used to add ”P at the gamma- phosphate position of ATP and the polynucleotide catalyses an exchange reaction with the 5’-terminal phosphate (Fig. 7).
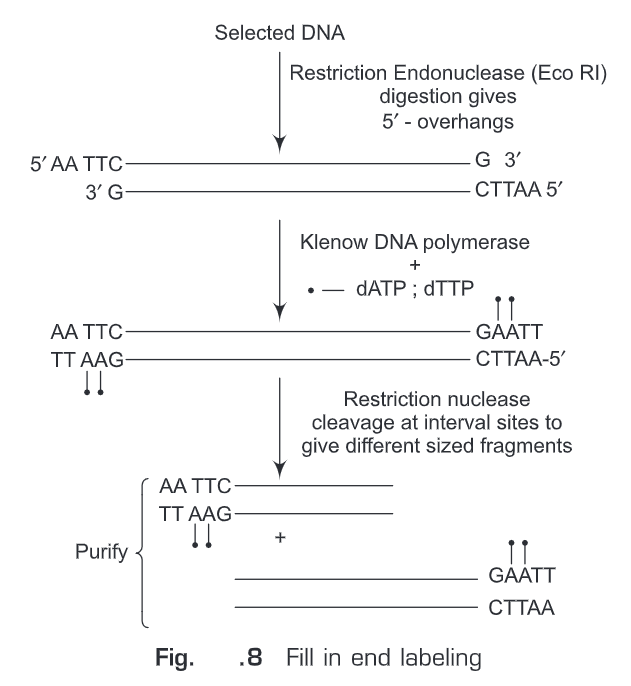
(ii) Fill-in end labeling (Fig. 8)— Initially, the selected DNA is digested with such restriction endonucleases that after digestion 5’-overhangs are produced (e.g., EcoRI). These 5’-overhangs will act as primer and polymerase activity of Klenow fragment (of E.coli DNA polymerase I) is used to fill in the recessed ends. Any one of the nucleotides provided in the reaction mixture is labeled. Hence, we get labeled oligonucleotide at 3’-end. Another specific restriction endonuclease can be used to cleave internal site so as to get fragments of different sizes which can be size fractionated. The advantages of this method are:
- Requires only a small amount of template
- Labeled probes can be used without purification
- Reaction can be scaled up indefinitely (if incubation time is increased to 1 hour)
(iii) Primer mediated 5’-end labeling – In primer mediated 5’-end labeling, a primer attached to a labeled group at its 5’-end is used in the normal PCR reaction. As PCR amplification proceeds, the 5’-end-labeled primer is incorporated in the amplified PCR product. The advantages of this method are:
- Requires only a small amount of template
- Produces more sensitive probes than end labeling
- Labeled probes can be used without purification
- Reaction can be scaled up indefinitely