Prions and transmissible spongiform encephalopathies
▶Transmissible spongiform encephalopathies
The transmissible spongiform encephalopathies (TSEs) are a unique suite of progressive neurodegenerative diseases that show both infectious and hereditable transmission characteristics. TSEs have been observed in various mammals including humans, and as yet no treatment is available. The pathogenic agent replicates in a number of tissues including spleen and liver, before appearing in high concentrations in the brain and central nervous system (CNS) towards the terminal stages of the disease.
Ultimately, severe degeneration of the brain and spinal cord lead to clinical signs and the condition is then invariably fatal within several months. Pathologically the disease is characterized by the appearance of abnormal protein deposits (amyloids) in the kidney, spleen, liver, and brain. Amyloids arise from the accumulation of various proteins, which take the form of plaques or fibrils. Amyloidosis is also a characteristic feature of Alzheimer’s disease, and although this condition is not transmissible, a prion-based pathology has not been totally eliminated. The protein deposits are insoluble and hence cytotoxic, leading to cell death and a sponge-like appearance of the brain, which gave the spongiform encephalopathies their name; microscopic holes can be visualized in thin sections of brain tissue taken at post mortem.
There is no conventional immune response to the agent, although the immune system plays an important part in the development of the disease before the agent gets into the CNS. Diagnosis of TSEs is difficult and for some time could only be made by immunohistochemical staining of the prion protein in tissue taken at post mortem, although there have been some examples of diagnosis based on detection of the prion protein in cerebrospinal fluid (CSF) or tonsillar tissue.
▶The prion agent
There has been much speculation as to the molecular nature of the infectious agents responsible for these disease conditions. In 1972 Stanley Prusiner proposed that these agents were not viruses but a unique class of infectious agent totally free of nucleic acid and consisting solely of protein. Prusiner coined the term prion (from proteinaceous infectious particle) to describe the agent and was awarded the Nobel Prize in 1997 for his work investigating prions and TSEs. Evidence for the proteinaceous, nucleic acid-free nature of the agent has come from a number of experiments in which the chemical and physical nature of the infectious agents has been examined.
They are resistant to heat inactivation at temperatures that would readily destroy DNA or RNA and are resistant to both ultraviolet light and ionizing radiation, treatments that normally damage microbial genomes. No susceptibility to DNAse, RNAse or enzymatic hydrolysis is detectable but the agents are sensitive to treatment with urea, SDS, phenol, and other protein-denaturing chemicals. These characteristics indicate that the prion has, as proposed, a proteinaceous character lacking an infectious or transmissible nucleic acid. Of course, all proteins are encoded by nucleic acids and the prion protein (PrP) is no exception, being encoded by a cellular gene, the Prnp gene. Found in all mammalian and bird genomes examined to date the protein is a glycoprotein of 208 amino acids that cycles between the endosomal and plasma membranes of various cell types, including neuronal cells. The biological function of the cellular protein, PrPc, is unknown although evidence suggests a role in transmembrane transport or signalling. Mice lacking this protein develop normally, suggesting redundancy in its function and critically such mice are also resistant to infection by disease-forming prion proteins (designated PrPsc was taken from the disease scrapie), indicating the requirement for the cellular protein in disease.
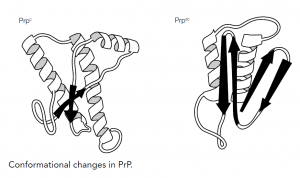
How do PrPc and PrPsc differ and how is the disease state achieved? The answer lies in the different conformations of the two proteins; the PrPc has a high proportion of alpha-helical structure but can undergo a conformational change to the pathogenic PrPsc form, which has reduced helical structure as regions refold into beta-sheets. This altered form of the protein shows increased resistance to proteases and reduced plasma membrane expression within the cell and, more importantly, increased self-aggregation, forming long fibrils of protein that eventually become insoluble, causing the cellular damage. How PrPc converts to PrPsc and whether the process is reversible is unclear, but there seems strong evidence that the presence of PrPsc ultimately converts more and more PrPc until the cell is destroyed.
The concept of prion protein conformational conversion supports the three disease routes observed with TSEs. Inheritance of a mutated Prnp gene can lead to familial (hereditary) disease, a spontaneous transcription error during gene expression would give sporadic disease and acquisition of the PrPsc isoform initiates the transmissible disease. As with conventional microbes, there are different strains of prion that appear to ‘breed true’ in terms of the characteristic symptoms they produce following infection of new hosts. For example, scrapie can be transmitted to both sheep and goats where, despite variations in the amino acid sequence of their PrP proteins, it displays identical disease characteristics. Likewise, some strains of BSE can be propagated by several animal species (each with their own different prion protein) and the three-dimensional conformation of the refolded PrPsc is identical regardless of the host species. These findings suggest that the final structure of PrPsc and its disease characteristics are independent of the host. By contrast, the transmission of scrapie (via contaminated feed) into cattle gives distinct characteristics and confirms that the contribution of infecting and host prion proteins is not yet understood.
In humans, there are two common versions (polymorphisms) of the prion protein that differ in single amino acid (valine or methionine at coding position 129). There are also rare mutant forms, many of which are associated with inherited susceptibility to prion disease. The polymorphic variation at position 129 does not itself lead to disease but influences the clinical characteristics of a disease-promoting mutation that can occur at position 178. Mutation of the normal aspartate to asparagine at this position leads to CJD if there is valine at position 129, but to fatal familial insomnia (FFI) if the individual possesses methionine at amino acid 129. Sheep likewise have several different forms of the prion protein linked to susceptibility, whereas cattle have two forms that do not appear to be associated with susceptibility to BSE.
▶Animal TSEs
Examples of TSEs in animals include scrapie in sheep and goats, transmissible mink encephalopathy (TME), feline spongiform encephalopathy (FSE), and bovine spongiform encephalopathy (BSE, labelled ‘mad cow disease’ by the media). Scrapie has been recognized as a distinct infection in sheep for over 250 years. A major investigation into its atiology followed the vaccination of sheep for louping-ill virus with formalin treated extracts of ovine lymphoid tissue, unknowingly contaminated with scrapie prions. Two years later, more than 1500 sheep developed scrapie from this vaccine. The scrapie agent has been extensively studied and experimentally transmitted to a range of laboratory animals, e.g. mice and hamsters.
Infected sheep show severe and progressive neurological symptoms, such as abnormal gait. The name derives from the fact that sheep with the disorder repeatedly scrape themselves against fences and posts. The natural mode of transmission between sheep is unclear although it is readily communicable and the placenta has been implicated as a source of prions, which could account for horizontal spread within flocks. In Iceland, scrapie-infected flocks of sheep were destroyed and the pastures left vacant for several years. However, the reintroduction of sheep from flocks known to be free of scrapie for many years eventually resulted in scrapie, suggesting that it was able to retain infectivity in the soil for several years. Sheep have also been infected by feedstuff contaminated with BSE, to which they are susceptible.
Bovine spongiform encephalopathy (BSE) appeared in Great Britain in 1986 as a previously unknown disease. Affected cattle showed altered behaviour and a staggering gait and post-mortem investigation revealed protease-resistant PrPsc in the brains of the cattle and the typical spongiform pathology. The origins of BSE suggest that cattle were infected with meat and bone meal (MBM) given as a nutritional supplement, contaminated with an unknown bovine TSE (research has shown that the BSE and scrapie prions are different, hence scrapie is not the ancestor of BSE).
MBM was initially prepared by rendering the offal of sheep and cattle using a process that involved steam treatment and hydrocarbon solvent extraction but in the late 1970s the solvent procedure was eliminated from the process, resulting in high concentrations of fat in the MBM. It is postulated that this high fat content protected prions in the sheep or beef offal from being completely inactivated by the steam. Since 1988 the practice of using dietary protein supplements for domestic animals derived from rendered sheep or cattle offal has been forbidden in the UK. Statistics argue that this food ban has been effective in controlling the epidemic and British beef has been reported as being free of BSE (June 2006).
▶Human TSEs
Prior to the mid-1990s there were four forms of human TSE identified; Creutzfeldt-Jakob disease (CJD), the rare, inherited fatal familial insomnia (FFI), Gerstmann-Straussler- Scheinker disease (GSS), and the now-extinct Kuru, which was the first human TSE to undergo extensive investigation. The first cases were recorded in the 1950s and occurred in male and female adolescents and adult women members of the Fore people in the highlands of New Guinea and at one point was the leading cause of death of women of the tribes. The Fore people practised ritual cannibalism as a rite of mourning for their dead, the women and children but not the adult men taking part in the ceremony. The disease demonstrated progressive loss of voluntary neuronal control followed by death within 1 year after the onset of symptoms.
As the cannibalistic ritual ceased so did transmission of the disease and although some individuals developed symptoms after an incubation period of up to 30 years (usually it was much shorter), the disease is now eradicated. Kuru demonstrated clearly that a human TSE could be acquired via the oral route. Patients with CJD present with a progressive subacute or chronic decline in cognitive or motor function associated with recurrent seizures. CJD is rare, with an annual worldwide incidence of approximately one per million population and can be familial (inherited; 10% of cases), or acquired sporadically or iatrogenically (cases have been recorded following corneal grafts, the administration of pituitary hormones or the use of electrodes contaminated with PrPsc).
In April 1996 a new variant of CJD (vCJD) was described in the UK. The disease has features that distinguish it from other forms of CJD including early age of onset (average 27 years as opposed to 65 for CJD), suggesting a shorter incubation period, a prolonged period of illness (average 13 months as opposed to 3 months for CJD), and a psychiatric presentation as opposed to neurological symptoms, with the absence of the typical electroencephalography (EEG) appearances of CJD. In terms of pathology, vCJD resembles
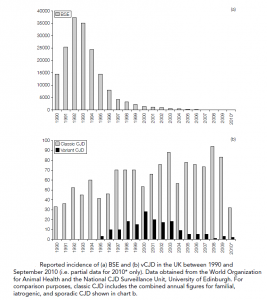
Kuru, particularly the type of amyloid plaque formation. Evidence has accumulated that the BSE prion has been transferred (most likely via the oral route) to humans and is responsible for vCJD. The source of the infected material is unknown, although BSE contaminated cattle products are a likely candidate. In 1989 the human consumption of specified bovine offal (brain, spleen, thymus, tonsil, and gut) was prohibited in the UK and in 1996 the ban was extended to sheep offal. Furthermore, the brain and CNS are now removed from all cattle at abattoirs before the distribution of meat. Up to September 2010, 118 cases of deaths as a result of vCJD have been confirmed, with a further 51 deaths likely to be due to this TSE (post-mortem confirmation pending). There is strong epidemiological evidence that the epidemic has peaked but it is worth considering that all but one case to date have occurred in individuals homozygous for valine at the position 129 polymorphism described above. Since all forms of the polymorphism support BSE in a mouse system, it is possible that there is an as-yet symptom-free cohort of infected individuals that are still incubating vCJD. Although it is considered highly unlikely, the possibility of human-to-human transmission remains.