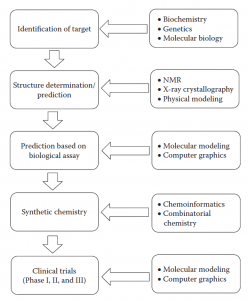
STRATEGIES IN COMPUTER-AIDED DRUG DESIGNING
1. Target Identification:
- Target Identification is a field that integrates different levels of information in drug – protein and protein – disease networks and involves an intricate network of databases and correlations across genomics , proteomics, transcriptomics, metabolomics, microbiome and pharmacogenomics.
- There are two approaches that can be used for target identification: the first is at the beginning (target-based or reverse chemical genetics) and the second is at the end (phenotype-based or forward chemical genetics) Biological screening.
- Target selection can be done in three ways: biochemical, genetic interaction, and computational inference.
- Biochemical affinity purification directly affects the target of small molecules through biochemical approaches.
- However, lead optimization is good at the availability of target structure information with the help of enzymes assays and other biochemical tests.
- Various DNA and RNA-based methods allow target identification via genetic or genomic methods for the functional analysis of protein targets in the controlled system.
- Numerous computational inference methods have also been reported for the predictions of targets of identified inhibitors using structural-based and (ligand-based) profiling methods.
2. Structure Prediction:
- In the early stages of the drug development process, researchers faced challenges due to little or no information on the Structure – Activity Relationship (SAR) information.
- The development of HTR techniques has now made it easier for chemists to develop the assay and screening of lead compounds.
- The selection of lead compounds is based on a set of compounds that have diversity in their physiochemical properties and are easier to select rather than randomly selected.
- The aim of these analyses is to select and test less compounds while obtaining as much information as possible about the data set.
- The selection of compounds to be tested is of primary importance as it has a strong impact on the number of compounds tested for research efficiency and also on the cost implications.
- Recent research on refinements and lead findings has been conducted on the rational design of drugs for the structural diversity of databases.
- Three-dimensional diversity enhancement databases were compared with hierarchical clustering and maximum dissimilarity methods.
- Rational selection and random approaches were compared for the performance of two-dimensional fingerprints as a validated molecular descriptor.
- This comparison leads to a choice of pharmacophores. The Pharmacophore model is a spatial arrangement of atoms or functional groups that explains the linkage of structurally diverse ligands to a common receptor site.
3. Biological assay-based target prediction:
- A bioassay or bioassay is defined as a test procedure that is used to estimate the concentration of a pharmaceutically active substance in a formulated product or bulk material.
- Of the different physical or chemical methods available, a bioassay provides exhaustive information on the biological activity of a substance.
- CADD-based drug target identification can be divided into drug-based similarity inference (DBSI), target-based similarity inference (TBSI) and network-based inference (NBI). The NBI method is considered to be the best method among them.
- These in vitro assays have shown that the five old drugs, namely montelukast, diclofenac, simvastatin, ketoconazole and itraconazole, exhibit polypharmacological properties on oestrogen receptors or dipeptidyl peptidase-IV.
- Target prediction may also be performed by receptor-based methods, such as reverse docking, which have also been used in drug-target (DT) binding affinity predictions. The availability of a 3D structure is the limitation of the target prediction.
REFERENCES:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5248982/
- https://www.researchgate.net/publication/8340229_Strategy_of_Computer-Aided_Drug_Design
ALSO READ :