Inhibitors of nucleic acid synthesis
The selectively synthetic antimicrobial agents interfere with the functions of DNA and RNA. Some are sulphonamides and diaminopyrimidines which achieve their effect indirectly by interrupting metabolic pathways that lead to the manufacture of nucleic acids; others, of which the quinolones and nitroimidazoles exert direct actions.
Sulphonamides
- It was earlier used for chemotherapy of bacterial infections. But its use has been reduced due to widespread acquired resistance and low potency compared to other drugs. Still, this is used in uncomplicated urinary infections caused by E. coli in domiciliary practice.
- They have also been used treating meningococcal meningitis and superficial eye infections
- They are still found in combination products with trimethoprim, pyrimethamine, and other diaminopyrimidines.
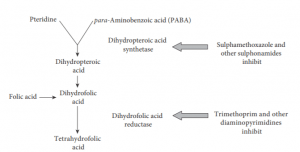
- Most bacteria synthesize folic acid and cannot take it up preformed from the environment. Mammalian cells, in contrast, use preformed folate and cannot make their own. Sulphonamides block an early stage in folate synthesis, thus leading to a failure to synthesize purine nucleotides and thymidine.
- Folic acid is an important chemical for the synthesis of bacterial DNA and RNA, is inhibited by sulfonamides
- Sulphonamides have a broad antibacterial spectrum, although the activity against enterococci, Pseudomonas aeruginosa, and anaerobes are poor.
- These agents relatively take time to almost several generations of bacterial growth are needed to deplete the folate pool before inhibition of growth occurs.
- Resistance emerges readily, and bacteria resistant to one sulphonamide are cross-resistant to the others.
Diaminopyrimidines
- Diaminopyrimidines are synthetic antimicrobial agents that interfere with folic acid production by inhibiting dihydrofolate reductase enzyme that generates tetrahydrofolate from metabolically inactive dihydrofolate
- Trimethoprim is the most important antibacterial agent which exhibits far greater affinity for the dihydrofolate reductase of bacteria than for the corresponding mammalian enzyme
- Since sulphonamides and trimethoprim act at different points in the same metabolic pathway,
they interact synergically and thus bacteria are inhibited by much lower concentrations of the combination than by either agent alone. - For this reason, trimethoprim and sulphonamides have been combined in therapeutic formulations such as trimethoprim-sulphamethoxazole
- Trimethoprim is active in low concentration against many common pathogenic bacteria,
although Ps. aeruginosa is a notable exception. - The drug is rapidly absorbed from the gut and excreted almost exclusively by the kidneys, with a plasma half-life of about 10 hours.
- The chief use for trimethoprim is in urinary tract infection (UTI).
Quinolones
- A new series of quinolones were synthesized, collectively known as fluoroquinolones.
- Quinolones are broad-spectrum antibiotics that are active against both Gram-positive and Gram-negative bacteria, including mycobacteria, and anaerobes.
- All antibacterial quinolones act against enzymes that are involved in maintaining the integrity of the supercoiled DNA helix during replication and transcription by causing breakage of bacterial chromosomes. Two enzymes are affected, DNA gyrase and topoisomerase IV (Topoisomerase Type II) so that these drugs have a dual-site of action.
- In Gram-negative bacilli, the main target is DNA gyrase, with topoisomerase IV as a secondary
site but, in Staphylococcus aureus and some other Gram-positive cocci, the situation is reversed. - Quinolones are generally well tolerated but rashes, gastrointestinal disturbances, genotoxicity, phototoxicity may occur.
- Quinolones are antagonists of GABA (gamma-aminobutyric acid) and should be used with caution in patients with epilepsy.
- These compounds affect the deposition of cartilage in experimental animals, and licensing authorities have cautioned against their use in children and pregnant women.
- All quinolones are well absorbed when taken by mouth and are more or less extensively metabolized in the body before being excreted into the urine.
- Nalidixic acid is a narrow-spectrum agent against Gram-negative bacteria (enteric bacteria), with the exception of Ps. aeruginosa, but Gram-positive organisms are usually resistant.
- The spectrum also includes certain problem organisms such as chlamydiae, legionellae, and some mycobacteria.
- Those fluoroquinolones, such as ciprofloxacin and ofloxacin, are active against both Gram-negative and Gram-positive pathogens and they are also active against the causative agent of tuberculosis, Mycobacterium tuberculosis.
- Fluoroquinolones are usually administered orally, although some, including ciprofloxacin,
ofloxacin, and levofloxacin, can also be given by injection. - The major route of excretion is usually renal, in the form of native compound or glucuronide and other metabolites.
Nitroimidazoles (anaerobic infections only)
- The imidazoles and its derivatives are remarkable synthetic antimicrobial agents known to cover bacteria, fungi, viruses, protozoa, and helminths—in fact, the whole antimicrobial spectrum.
- The members of this family used as antibacterial agents are 5 nitroimidazoles, of which metronidazole is best known.
- Related 5-nitroimidazoles include tinidazole and ornidazole, both of which share the properties of metronidazole but have longer plasma half-lives.
- Metronidazole was originally used for the treatment of giardiasis caused by Giardia lamblia that causes gastrointestinal disease and subsequently for two other protozoal infections—amoebiasis and trichomoniasis.
- These drugs are considered to damage DNA but the exact mechanism is yet unknown.
- Anaerobic bacteria are commonly incriminated in gingivitis, and it was subsequently shown that metronidazole possesses potent antibacterial activity against strict anaerobes and also some microaerophilic bacteria, including Entamoeba histolytica, Gardnerella vaginalis and Helicobacter pylori.
- Metronidazole is so effective against anaerobic bacteria, and resistance to it is so uncommon,
that it is now the drug of choice for the treatment of anaerobic infections. - It is also commonly used for prophylaxis in some surgical procedures in which postoperative anaerobic infection is a frequent complication.
- The basis of the selective activity against anaerobes resides in the fact that a reduction product
is produced intracellularly at the low redox values attainable by anaerobes but not by aerobes. - The 5-nitroimidazoles are generally free from serious side effects, although gastrointestinal
upset is common and ingestion of alcohol induces a disulphiram-like reaction. - Other imidazole derivatives include clotrimazole, miconazole and econazole, all of which possess a broad antimycotic spectrum with some antibacterial activity and are used topically
Nitrofurans (nitrofurantoin)
- A number of nitrofuran derivatives have attracted attention over the years, among which Nitrofurantoin and Nitrofurazone are most important.
- The nitrofurans have remained clinically useful against a wide spectrum of Gram-positive and Gram-negative bacteria, including many strains of common urinary tract pathogens
- Nitrofurantoin use is restricted to the treatment of lower urinary tract infection since it is rapidly excreted into urine after oral absorption, and the small amount that finds its way into tissues is inactivated there while Nitrofurazone is primarily used as a topical antibacterial agent in burns and skin grafts and recently was approved for the prophylaxis of CAUTI (catheter-associated urinary tract infection)
- It appears that the nitrofurans act by inhibiting bacterial enzymes involved in DNA and RNA synthesis, carbohydrate metabolism, and other metabolic enzyme proteins.
- When taken orally for urinary tract infection (UTI), nitrofurantoin is rapidly metabolised in renal tissues and is rapidly excreted into the urine via glomerular filtration and tubular secretion.
Rifamycins (rifampicin)
- The clinically useful rifamycins, of which rifampicin is the most important, are semi-synthetic derivatives of rifamycin B, one of a group of structurally complex antibiotics produced by Streptomyces mediterranei.
- Rifamycins inhibit RNA polymerase of most bacterial genera. These compounds interfere with mRNA formation by binding to the β-subunit of DNA-dependent RNA polymerase.
- Rifampicin remains part of combination therapy for treating tuberculosis (TB), and for treating Gram-positive prosthetic joint and valve infections, in which biofilms are prominent.
- Resistance readily arises by mutations in the subunit. For this reason, the drugs are normally used in combination with other agents.
Rifampicin
- Rifampicin has potent activity against a variety of pathogens, including mycobacteria, Gram-positive cocci (notably staphylococci and streptococci), Clostridium difficile, and has activity against select Gram-negative pathogens Neiserria meningitides, N. gonorrhoeae, and Hemophilus influenza.
- Rifampicin is one of the most effective weapons against two major mycobacterial scourges of
mankind: tuberculosis and leprosy. - Rifampicin is now often used in combination with other drugs in Legionnaires’ disease and staphylococcal prosthetic device infections.
- It is also used as a single agent to eliminate meningococci from the throats of carriers and for the protection of close contacts of meningococcal and Haemophilus influenzae type b disease.
- Rifampicin is well absorbed by the oral route, although it may also be given by intravenous infusion.
- Hepatotoxicity is well recognized and the antibiotic induces hepatic enzymes, thus leading to self-potentiation of excretion and the antagonism of some other drugs handled by the liver, including oral contraceptives.
- Rifampicin is strongly pigmented and led to the production of red urine and other bodily secretions; contact lenses may become discolored.
- Two other rifamycin derivatives (rifabutin and rifapentine) have a limited role in the treatment
of infections caused by organisms of the Mycobacterium avium complex, which often cause disseminated disease in patients with cancer or acquired immune deficiency syndrome (AIDS).
Chloramphenicol
- Chloramphenicol was initially derived from Streptomyces and has since been fully synthesized. It has a wide activity spectrum but exerts a bacteriostatic influence.
- It has antirickettsial function and the larger viruses are inhibitory.
- Chloramphenicol diffuses through the bacterial cell wall and reversibly binds to the bacterial 50S ribosomal subunit.
- Unfortunately, for a proportion of patients, aplastic anemia, which is dose-related, can result from treatment. Therefore, it should not be administered for mild diseases and its use should be limited to situations where there is no suitable solution, such as typhoid fever that is resistant to other antibiotics.
- An enzyme, chloramphenicol acetyltransferase, can be produced by certain bacteria, which acetylates the hydroxyl groups in the side-chain of the antibiotic to initially generate 3acetoxychloramphenicol and eventually 1,3diacetoxychloramphenicol, which lacks antibacterial action.
- A significant result may be the design of fluorinated chloramphenicol derivatives that are not acetylated by this enzyme, although toxicity might be a concern.
- The antibiotic is delivered orally as tasteless palmitate; this is hydrolyzed in the gastrointestinal tract into chloramphenicol.
- In the parenteral formulation, the highly water-soluble chloramphenicol sodium succinate is used and hence serves as a pro-drug. Thiamphenicol, a semisynthetic chloramphenicol analog, is believed to be less toxic than chloramphenicol.
References
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4354722/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6836748/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3862438/
- https://pubmed.ncbi.nlm.nih.gov/11293646/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4930915/
- https://pubchem.ncbi.nlm.nih.gov/compound/Chloramphenicol