Prenatal Diagnosis: Overview
INTRODUCTION
One of the most important threats for human’s health is the genetic diseases. Genetic disease is a disorder caused by genetic factors (especially abnormalities) in the human genome. The four main types of genetic diseases are:
- Single-gene/monogenic Genetic Diseases
- Multi-factorial/Polygonic Genetic Diseases
- Chromosomal Genetic Diseases
- Mitochondrial Genetic Diseases
- Single-gene (Mendelian or monogenic): The starting point is a mutation/change in the DNA sequence of one gene. Genes code for proteins. When there is a mutation in the gene, the resulting protein can no longer carry out its normal function, therefore, results in a disorder/ disease. Around 6000 single gene disorders are known. Some examples are cystic fibrosis, sickle cell anemia, Marfan syndrome, Huntington’s disease, etc.
- Multi-factorial/Polygonic/Complex Genetic Diseases: Multi-factorial genetic disease is caused by environmental factors and mutations in more than one gene. These abnormalities are also difficult to analyze. Some of the most common chronic disorders are multi-factorial. For examples—heart disease, high blood pressure, Alzheimer’s disease, arthritis, diabetes, cancer, and obesity.
- Chromosomal Genetic Diseases: Chromosomes are carriers of genetic material (composed from genes). Abnormalities in chromosome structure and number of the chromosomes can lead to most dangerous genetic disorders. This type of disorders can be detected by microscope examination. Down syndrome or trisomy 21 is a common disorder that occurs when a person has three copies of chromosome 21.
- Mitochondrial Genetic Diseases: It is a rare type of genetic disorder and is caused by mutations in the non-chromosomal DNA of mitochondria.
DIAGNOSIS OF GENETIC DISEASES AT DNA LEVEL
Earlier diagnosis of genetic diseases was based on biochemical assays which only reported the presence or absence of a gene product. But DNA based tests determine the existence of specific gene mutations. Test at DNA level use expression for the detection of a mutant gene. Diagnosis of genetic diseases at DNA level can be used for the identification of.
Prenatal Diagnosis
Prenatal diagnostic testing involves the testing of the fetus prenatal (before birth) to determine whether it has a certain hereditary or spontaneous genetic disorder. Therefore, prenatal diagnosis employs a variety of techniques to determine the health and condition of an unborn fetus. Prenatal diagnosis has been a major focus for genetic disease diagnosis.
The principal aim of prenatal diagnosisis to give information to the families that whether they or their offspring are at risk or not. Approximately, 34% of all births are associated with genetic disorders, mental retardation or congenital defects. But the prenatal diagnosis of such cases has enabled obestricians to avoid birth of such fetus. There is a number of non-invasive and invasive techniques available for prenatal diagnosis. Each of them can be applied only during specific time periods during the pregnancy for greatest utility. Prenatal diagnosis includes both screening and diagnostic methods (techniques).
Prenatal screening for genetic diseases is performed when
- Positive maternal serum marker
- Parental chromosomal rearrangement
- Advanced maternal age (more than 35 years)
- Family history of genetic diseases
- History of unexplained fetus loss
- Prenatal Diagnostic Methods are divided into two types:
Analysis of fetal tissue
- Amniocentesis— This is an invasive procedure. A sample of amniotic fluid (surrounding the fetus) is collected and then analyzed for single gene disorders and chromosomal based abnormalities. Amniocytes are cultured to increase their number and then cytogenetic studies are carried out. Level of alpha-fetoprotein is used to detect the presence of neural tube defect. It is performed after 15 weeks of pregnancy and it takes around 10-12 days for cytogenetic studies.
- Chorionic villus sampling (CVS)—This is an invasive procedure. CVS is performed by aspirating fetal trophoblastic tissue (chorionic villi). CVS is preferred for chromosomal and DNA based diagnosis. It is performed between 10-12 weeks of pregnancy. CVS results are available earlier than amniocentesis.
- Percutaneous umbilical blood sampling (PUBS) or Cordocentesis— PUBS is the preferred method towards the end of pregnancy when ultrasonography has detected abnormalities in the fetus. PUBS is used for rapid chromosome analysis. A needle is inserted through the abdominal wall into the umbilical cord. Fetus blood sample is aspirated and analyzed. Applications of PUBS are- diagnosis of hematological diseases, cytogenetic analysis of fetuses with structural anomalies, etc.
Visualization
- Ultrasonography—This is a non-invasive procedure that is harmless to both the fetus and the mother. A transducer is placed on the mother’s abdomen. This transducer sends pulsed sound waves through the fetus. The fetus in turn reflects these sound waves in patterns corresponding to the tissue density. The reflected waves are displayed on the monitor giving the visualization of the fetus.
Prenatal diagnosed (amniocentesis or CVS) disorders are:
- Lesch-Nyhan syndrome
- Tay sachs disease
- Galactosemia
- Maple syrup urine disease, etc
Prenatal diagnosed (ultrasonography) disorders are:
- Congenital heart disease
- Intrauterine growth retardation
- Cystic kidneys
- Limb reduction defects, etc
Prenatal diagnosis, therefore, provides an opportunity to avoid the birth of mentally and physically handicapped babies and hence offers the potential for improvement in the health of human race.
Diagnosis before Onset of Symptoms
Diagnosis of a disease before the onset of symptoms can be done by analyzing the wild genotype and then comparing it with the mutant genotype by using the Genotyping with fluorescence labeled PCR primers method. Fluorescence resonance energy transfer is one of the most important tools for single nucleotide genotyping. We can also detect a single base mutation by this method.
Genotyping with fluorescence labeled PCR primers method—Genotyping with fluorescence labeled PCR primers is a non-radioactive detection system. This method is based upon allele- specific primer extension labeled with fluorescent dyes. It involves the labeling of PCR primers with different fluorescent dyes on the basis of which different genotype are identified. To distinguish between wild and mutant genotype, two different primers labeled with different fluorescent dyes are used.
One of the primers is complementary to the wild genotype and labeled at its 5’ end with rhodamine and the other primer is complementary to the mutant genotype and is labeled with flourescein at its 5’end. PCR amplification is programmed in this case by using an unlabelled third primer complementary to the opposite strand.
Then PCR amplification is carried out by using these labeled primers. Amplification will take place only if the primers are exactly complementary to the target genotype. Therefore, either the wild genotype or the mutant genotype or the both will be amplified depending upon the annealing of the primers with the target DNA and the results will be:
- The amplified reaction mixture will fluorescence red if the individual is homozygous for the wild genotype
Wild genotype (Homozygous, +/+)
↓
Add primers P1 (labeled with rhodamine) and P2
↓
PCR
↓
Amplified product
↓
Red Fluorescence
- The amplified reaction mixture will fluorescence green if the individual is homozygous for the mutant genotype
Mutant genotype (Homozygous, -/-)
↓
Add primers P3 (labeled with flourescein) and P2
↓
PCR
↓
Amplified product
↓
Green Fluorescence
- The amplified reaction mixture will fluorescence yellow if the individual is heterozygous and has both wild and mutant genotype
Mutant genotype (Heterozygous, +/-)
↓
Add primers P1, P3 (labeled with rhodamine and fluorescein respectively) and P2
↓
PCR
↓
Amplified product
↓
Yellow Fluorescence
Carriers of Hereditary Disorders
The best example for carriers of hereditary disorders is sickle cell anemia.
Sickle cell anemia:
- Genetic disorder life-long blood disorder
- Single nucleotide mutation in the B-chain of the hemoglobin molecule
- Valine changes to Glutamic acid (6th amino acid)
- Sickle-shaped RBC (Sickle-shaped” means that the red blood cells are shaped like a “C.”)
- Sickle-shaped cells don’t move easily through the blood vessels. They’re stiff and sticky and tend to form clumps and get stuck in the blood vessels
- Biological ramifications are:
- Severe anemia – the inability of the mutated (aped RBCs) to carry sufficient oxygen
- Damage to brain, lungs, heart, and other organs
- Short life expectancy (homozygous for disease)
- Carriers (heterozygous for disease) individuals are normal until exposed to extreme conditions (High altitude, low oxygen, etc)
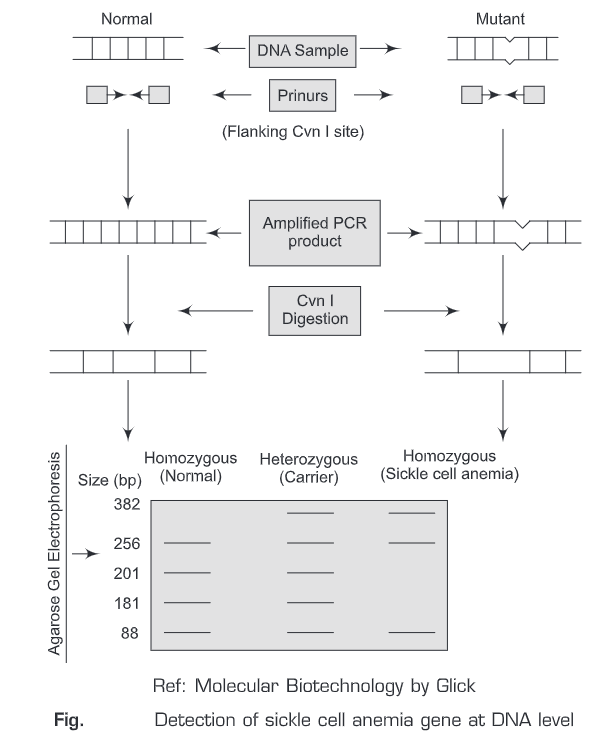
Detection of sickle cell anemia gene at DNA level: The single nucleotide mutation that leads to sickle cells anemia removes (abolishes) a restriction endonuclease site (CvnI). CvnI restriction endonuclease cleaves between Cytosine and Thymine residues when it recognizes the sequence CCTNAGG.
Normal gene sequence – CCTGAGG (Wild genotype)
Sickle cell anemia gene -CCTGTGG (Mutant genotype)
This forms the basis for the detection of sickle cell anemia gene. In case of wild genotype, there are three Cvn I restriction sites and in sickle cell anemia gene there are two restriction sites. Therefore, we can differentiate between homozygous (normal and sickle cell anemia) and heterozygous (carrier) individual on the basis of the number of bands obtained after Cvn I digestion. For wild (homozygous) individual, we get four bands; for sickle cell anemia (homozygous), we get three bands; and for sickle cell anemia (heterozygous/carrier), we get five bands.
(i) Normal gene sequence
Wild genotype
↓
Add primers flanking the Cvn I restriction endonuclease site
↓
PCR
↓
Amplified normal sequence
↓
Cvn I digestion (3 cleavage sites)
↓
Separation on gel electrophoresis (four bands)
↓
Visualization by ethidium bromide staining
(ii) Sickle cell anemia gene (homozygous)
Mutant genotype
↓
Add primers flanking the Cvn I restriction endonuclease site
↓
PCR
↓
Amplified sickle cell anemia sequence
↓
Cvn I digestion (2 cleavage sites)
↓
Separation on gel electrophoresis (three bands)
↓
Visualization by ethidium bromide staining
(iii) Sickle cell anemia gene (heterozygous)
Mutant genotype
Add primers flanking the Cvn I restriction endonuclease site
↓
PCR
↓
Amplified sickle cell anemia sequence
↓
Cvn I digestion (4 cleavage sites)
↓
Separation on gel electrophoresis (five bands)
↓
Visualization by ethidium bromide staining
MUTATIONS AT DIFFERENT SITES WITHIN ONE GENE
Mutations are changes in the DNA sequence caused by radiation, viruses, transposons and mutagenic chemicals. A condition caused by mutations in one or more genes is called a genetic disorder. Genetic diseases may be because of a single nucleotide mutation in a gene or more than two mutations in the same (intra-genic) or different (inter-genic) genes.
Sometimes, a number of different intra-genic mutations can lead to the same form of genetic disorder. For example, thalassemia. This is the most familiar type of thalassemia. It involves decreased production of normal adult hemoglobin (Hb A), (All hemoglobin consists of two parts: heme and globin). The globin part of Hb A has 4 protein sections called polypeptide chains.
Two of these chains are identical and are designated the alpha chains. The other two chains are also identical to one another but differ from the alpha chains and are termed the beta chains. In persons with beta-thalassemia, there is reduced or absent production of beta-globin chains. In people with beta-thalassemia, low levels of hemoglobin lead to a lack of oxygen in many parts of the body. Affected individuals have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications.
There are approximately eight mutation sites in beta-globin chain that leads to β-thalassemia. Homozygous individuals for β-thalassemia (homozygous for one or more mutation sites) have to take regular blood transfusions and other treatments also. Heterozygous (carriers) individuals have only a mild form of anemia. As mutation at any one or more than one site in the beta-globin chain can lead to β thalassemia, therefore we need to perform eight such tests (or a single test to diagnose all the eight mutation sites) to detect the exact mutation site. But such tests will not be cost-effective.
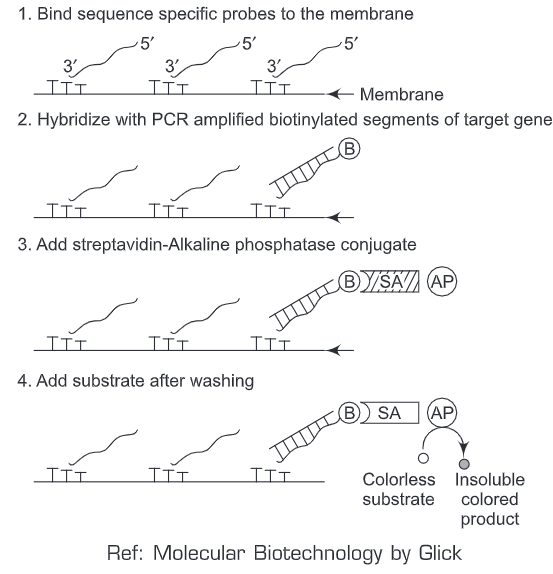
Therefore, a PCR-hybridization method has been devised to screen the different mutation sites within the same gene by using one reaction assay system. We’ll discuss PCR-hybridization method in context to screening of different mutation sites for β-thalassemia. Firstly, we synthesize a set of eight sequence (mutation) specific oligonucleotide probes (P1-P8) such that each one of the synthesized oligonucleotide perfectly matches with a segment of target gene that corresponds to a particular mutation. A poly dT (Thymidine homopolymer, = 400 nucleotides) tail is added to 3’end of all the eight oligonucleotids. This poly dT tail helps in the physical attachment of the oligonucleotide to a predefined discrete spot on the nylon membrane and the rest of the oligonucleotide is free for further hybridization step.
All the eight sequence (mutation) specific oligonucleotide probes (P1-P8) are bound to the nylon membrane. Simultaneously, DNA (segments) test sample is amplified by PCR using primers such that one of each of the pairs is labeled with biotin (B) at the 5’end. Then conditions that allow the perfect hybridization of amplified DNA sequence (biotin labeled) and membrane bound probes are applied. The amplified DNA sequence (biotin labeled) will bind to that specific probe (say P2) only which has the complementary sequence specific to a particular mutation.
Then, streptavidin (SA) conjugated with alkaline phosphatase, AP (or horse radish peroxidase or urease) is added to the hybridization reaction mixture. Streptavidin- alkaline phosphatase conjugate will bind to biotin labeled amplified DNA sequence only (as streptavidin has high affinity for biotin). After a particular duration of time when hybridization has occurred, the membrane is given a washing to remove any unbound material. Then a colorless substrate is added to the hybridized reaction mixture. A colored spot will appear at the position (P2) where the amplified DNA sequence matches with a specific (mutation at site 2) oligonucleotide probe. Thus, we can detect the exact mutation site (amongst the entire eight mutation site) in a single gene by using a single filter assay.